Overview
De novo genome assembly is the process of splicing DNA fragments into contiguous segments (overlapping clusters) representing the chromosomes of an organism. Accurate, complete and contiguous genome assemblies are essential for identifying important structural and functional elements of the genome and for recognizing genetic variation. However, the short read lengths produced by conventional sequencing technologies result in highly fragmented and incomplete assemblies. Short read lengths fail to span important genomic regions, such as repetitive sequences and structural variants, causing them to assemble incorrectly. With the development of long-read sequencing technologies, Pacific Biosciences Single Molecule Real-Time (SMRT) Sequencing and Oxford Nanopore Technologies can provide long and ultra-long sequencing reads that can easily traverse the most repetitive regions of the human genome, enabling the generation of highly contiguous genome assemblies. However, potential differences in their chemistry and sequence detection methods can affect their read lengths, base accuracy, and throughput.
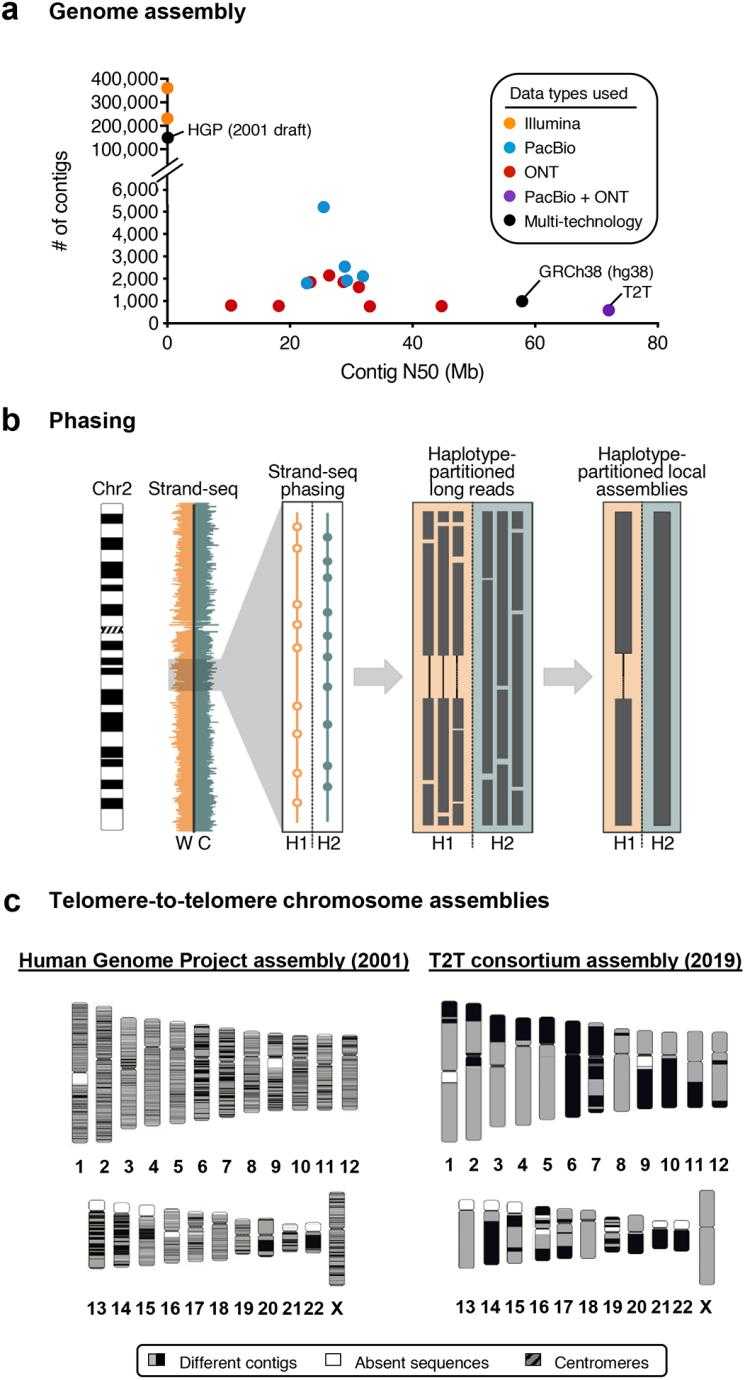
Long-read data improves genome assembly. (Logsdon GA et al., 2020)
Workflow of Long-read Sequencing for Generating Genome Assemblies
(1) Sample Preparation and Library Construction
Starting with a pure DNA sample, the first step is to fragment the DNA to the desired size. The advantage of long-read, long-sequencing technology is that it can handle very long DNA fragments, often spanning tens to hundreds of bases. Next comes the preparation of the sequencing library, which involves attaching specific junctions to these fragments. Companies such as Oxford Nanopore offer specialized kits, such as the Ultra-Long DNA Sequencing Kit, that facilitate the sequencing of very long fragments.
(2) Sequencing Run
Once the library is ready, it is loaded onto the sequencing equipment. The sequencing process relies on detecting changes in the electrical current as the DNA strand passes through the nanopore. This real-time detection is translated into a nucleotide sequence. Notably, Oxford Nanopore’s MinION and PromethION devices are capable of generating tens of kb-long reads, recording over 4 Mb.
(3) Data Analysis and Genome Assembly
The raw data (often referred to as “fast5” or “fastq” files) undergoes base recognition to convert electrical signals into nucleotide sequences. After this, a number of bioinformatics tools facilitate quality control, read matching and genome assembly. Long reads significantly reduce the complexity in short read assembly, especially when dealing with repetitive regions. Many softwares suite is specifically optimized for long read data for generating contiguous and high-quality genome assemblies.
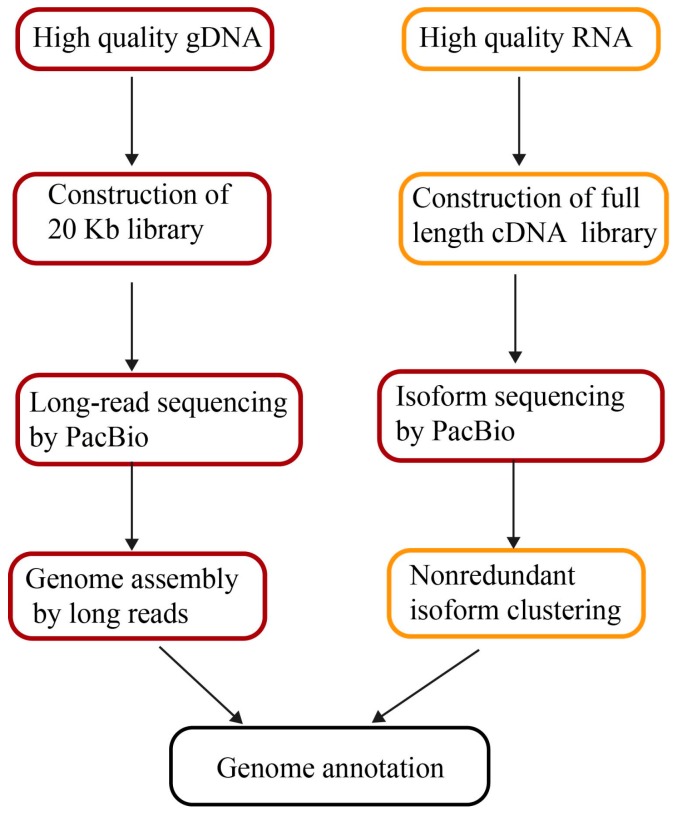
The pipeline of genome assembly and annotation by long reads. (Li C et al., 2017)
Applications of Long-read Sequencing for Generating Genome Assemblies
Resolving complex genomic regions
One of the persistent challenges in genomics is the accurate assembly of regions filled with repetitive sequences, structural variants, and GC-rich regions. Long sequencing reads can span these challenging regions, providing previously unattainable resolution. For example, sequencing the banana genome using Oxford nanopore technology showed fewer overlapping clusters and more complete chromosome reconstruction than short read-length methods.
Direct detection of modified bases
In addition to sequencing, the long-read method can detect base modifications such as methylation due to its direct sequencing approach. This provides the dual advantage of deducing nucleotide sequences and simultaneously understanding epigenetic modifications without additional experimentation.
Sequencing smaller microbial genomes in a single read
An incredible application of long-read sequencing is its ability to sequence smaller microbial genomes in a single read length. This completely eliminates the assembly process. For microbial researchers, this means faster insights and a deeper understanding of microbial diversity.
Crop improvement and breeding programs
In agriculture, access to high-quality reference genomes can significantly accelerate breeding programs. For example, scientists at KeyGene in the Netherlands have generated the most contiguous lettuce genome assembled to date using long-read sequencing. Such detailed genomic information can help select for important breeding traits and thus bring improved crop varieties to market faster.
Exploring evolutionary and symbiotic relationships
The depth and breadth of long-read sequencing also allows researchers to study the genome evolution of unique organisms. A prime example is the sequencing of lichen fungi, which are an integral part of many terrestrial ecosystems. Through long-read long sequencing, a more contiguous genome assembly is generated, leading to a better understanding of the symbiotic relationships of these fungi and their role in the environment.
References
- Logsdon, Glennis A., Mitchell R. Vollger, and Evan E. Eichler. “Long-read human genome sequencing and its applications.” Nature Reviews Genetics 21.10 (2020): 597-614.
- Li, Changsheng, et al. “Genome sequencing and assembly by long reads in plants.” Genes 9.1 (2017): 6.
